CAR-TOr TCR-T is discouraged? Repeated infusion of specific CAR or TCR mRNA- nanoparticles -T cells can alleviate the disease.
Engineering chimeric antigen receptor (CAR) or T cell receptor (TCR) is helpful to create disease-specific T cells for targeted therapy, but the cost and rigor of manufacturing engineering T cells in vitro may be prohibitive, so programming T cells in vivo may be a feasible alternative. An injectable nanocarrier is reported here, which delivers in vitro transcribed (IVT) CAR or TCR mRNA for instantaneous reprogramming of T cells to recognize disease-related antigens. In mouse models of human leukemia, prostate cancer and hepatitis B-induced hepatocellular carcinoma, repeated infusion of these polymer nanocarriers can induce enough host T cells to express tumor-specific CAR or virus-specific TCR, thus leading to disease regression, and the level is similar to that of in vitro engineered lymphocytes. Considering that they are easy to manufacture, distribute and manage, these nanocarriers and related platforms may become treatments for many diseases.
Adoptive T cell therapy is an effective method to treat cancer or infectious pathogens by genetic modification of T cells obtained from patients or donors, which has been supported by a large number of clinical trials and showed impressive results. However, the complexity and high cost of making customized T cell products for each patient, rather than preparing drugs in batches in a standardized form, make it difficult to compete with first-line treatment schemes such as small molecule drugs or monoclonal antibodies. At present, most CAR-T and TCR-T cells are manufactured through complicated processes, including: (i) white blood cell separation, and T cells are extracted from patients who are connected to the separator through two venous catheters for several hours. This is uncomfortable for patients, will generate a lot of money costs, and may eventually limit the large-scale adoption of autologous T cells; (ii) activation and transduction of T cells; (iii) expanding the transduced T cells in a tissue culture medium supplemented with cytokines for about 2 weeks; (iv) T cells are washed and concentrated before administration. For T cell products produced in central facilities and transported to remote treatment centers, cells must be frozen; (v) Every batch of CAR-T products needs to be tested for quality control and release. The whole process must be carried out under GMP-compliant environmental control conditions, and the maintenance and operation costs are very high. Because every CAR-T product is made of the starting material (T cells) of the patient to be treated, there is no economies of scale.
In vitro transcription (IVT)mRNA has become a subversive new drug, which can be used to directly encode protein related to treatment in vivo. The synthesized mRNA molecules can be designed and operated quickly, and mass-produced relatively economically and efficiently. In the past few decades, scientists have learned how to optimize mRNA from pharmacology and immunology, so that it can be used in clinical applications more like drugs.
Here, we explore using mRNA as an injectable drug to reprogram circulating T lymphocytes to express disease-specific receptors instantaneously, thus bypassing the need to extract and culture lymphocytes from patients (Figure 1). In order to protect the therapeutic load and accurately target it to T cells, biodegradable polymer nanocarriers were developed. First, it was proved in vitro that the application of a single nanoparticle can use CD19-specific 1928z CAR(FDA approved for the treatment of B-cell lymphoma) or HBcore18-27 TCR for HBV core antigen (currently in the phase I study for the treatment of patients with HBV-related hepatocellular carcinoma; NCT03634683) routinely transfected more than 70% of cultured T cells. T cells transfected with nanoparticles instantaneously express these CAR transgenes or TCR transgenes on their surfaces for an average of 7 days. In-situ xenotransplantation mouse models of lymphoma, prostate cancer and HBV-induced hepatocellular carcinoma have proved that mRNA particles encoding CAR or TCR can genetically reprogram circulating T cells when given regularly, so as to induce similar therapeutic effects to traditional adoptive transfer T cells transduced by virus in vitro.
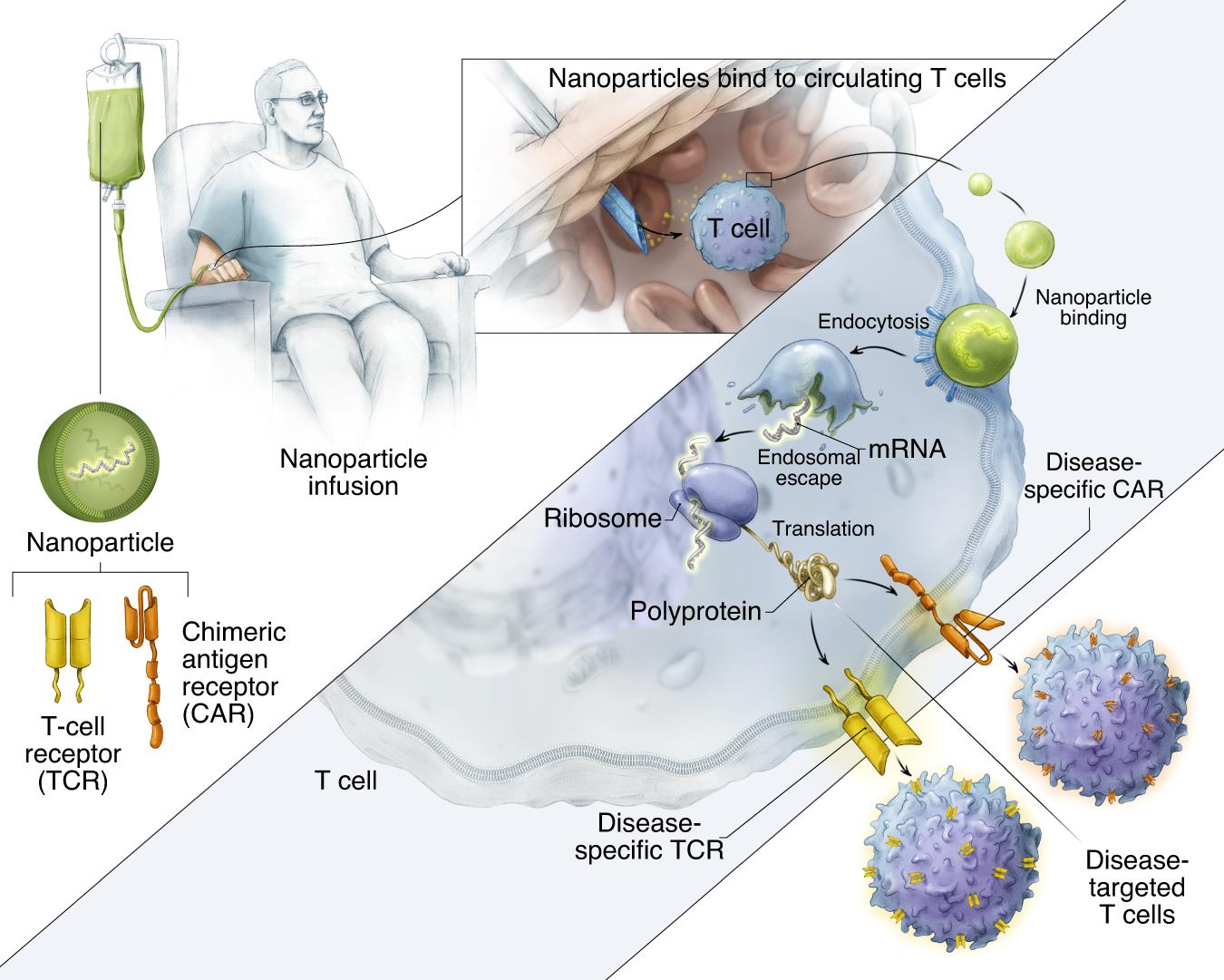
Fig. 1 is a schematic diagram of how to reprogram T cells in situ with IVT mRNA carried by polymer nanoparticles to express disease-specific CARs or TCRs. The surface of these particles is covered with ligands targeting cytotoxic T cells, so once they are injected into the circulation of patients, the transgene they carry can be transferred to lymphocytes, and the cells can be instantly programmed to express their disease-specific CAR or TCR on their surfaces.
At present, the insertion of CAR or TCR into lymphocytes by gene transfer is carried out in a special manufacturing workshop outside the patient’s body, but the process of transferring cells to and from the clean room and the gene transfer procedure itself are labor-intensive, costly and time-consuming. If engineered T-cell therapy reaches its promise of expanding to different populations of various cancer types, the economic and manufacturing challenges may increase.
Here, it is proved that mRNA nano-drugs can achieve effective cell therapy without side effects through the convenience of ready-made drugs. Just like traditional medicine, with this new treatment, patients can easily re-administer medicine as long as they need it.
CAROr TCR gene mRNA nanocarrier transfecting T cells.
In order to deliver IVT mRNA encoding disease-specific receptor gene to human lymphocytes, a biodegradable poly-β-amino ester (PBAE) polymer preparation was used as the carrier matrix (Figure 2a). The PBAE-447 polymer used in the research to concentrate mRNA into nanoparticles was originally developed by Jordan Green Laboratory of Johns Hopkins University. In the past ten years, the key features of PBAE have been widely described. PBAEs escape endosomes by protonation at low pH value, and osmotic pressure accumulates due to buffering, which leads to endosomes destruction. Qualcomm combinatorial library screening of PBAEs for nucleic acid delivery showed that the existence of tertiary amine improved the buffering capacity at low pH and promoted endosome escape. The ester bond in the main chain structure of PBAEs is hydrolyzed in aqueous solution, which makes the toxicity of PBAEs lower than that of other non-degradable cationic polymers, such as PEI, which is widely studied as a nucleic acid delivery carrier. Cationic PBAE self-assembled with anionic nucleic acid into nano-complex through electrostatic interaction (Figure 2b). By coupling anti-CD8 antibody to polyglutamic acid (PGA), the particles were electrostatically adsorbed to form a conjugate, which made the particles have cell targeting. The mRNA nanocarriers thus obtained can be freeze-dried for long-term storage. Before use, the granules were hydrated within a few seconds after adding sterile water to restore their original concentration. Particle tracking analysis (NanoSight NS300, Malven Panalytical) to characterize particles produced in ten independent batches (fig. 2c). The results show that the average particle size of PbAE/PGA-anti-CD8 nanoparticles is 106.9±7.2nm. The zeta potential is 4 2, and the encapsulation efficiency of mRNA detected by Qubit RNA HS kit is 90.9 6.2%. When the used nanoparticle formula PBAE: mRNA is 60: 1,
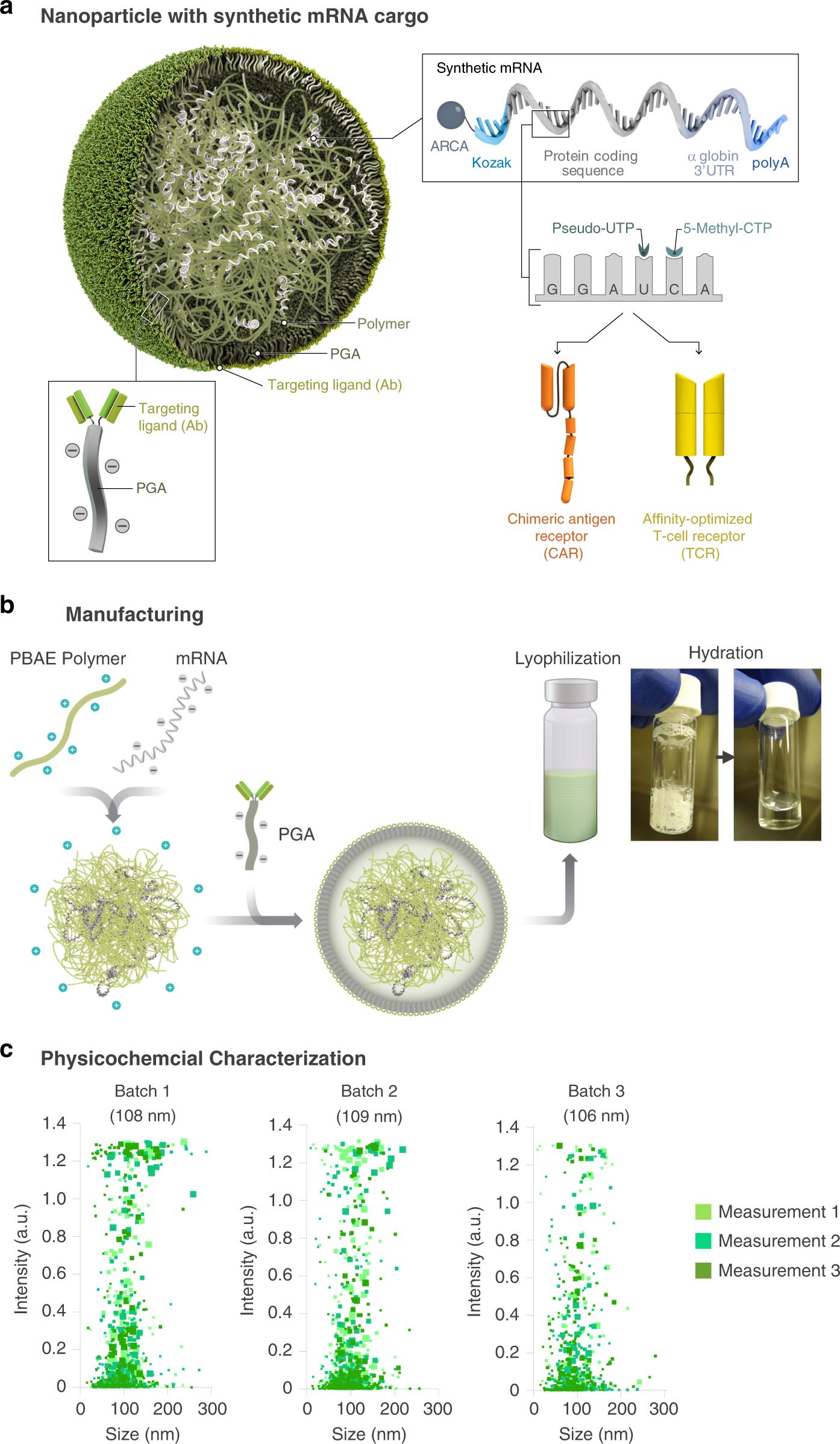
Fig. 2 Design and manufacture of lymphocyte programming nanoparticles. A: Schematic diagram of T cell targeting IVT mRNA nanocarriers used in the experiment. In order to create a reagent that can modify primary T lymphocytes simply by contact (which is difficult to achieve by non-viral infection methods), polymer nanoparticles composed of four functional components are designed: (i) surface-anchored targeting ligands, which selectively bind nanoparticles to T cells and initiate rapid receptor-induced endocytosis to internalize them. Anti-CD8 antibody was used in the experiment. (ii) negatively charged coating, which shields the nanoparticles by reducing the surface charge of the nanoparticles, so as to minimize off-target binding. Because it has been widely used in drug delivery platform, PGA was chosen to realize this. (iii) a carrier matrix, which aggregates and prevents nucleic acids from being degraded by enzymes when they are in the endocytosis, but releases them once the particles are transported into the cytoplasm, thus enabling the translation of the encoded protein. Therefore, a biodegradable poly (β-amino ester) (PBAE) polymer formula is used, and its half-life in water is between 1 and 7 hours. (iv) Nucleic acid (IVT mRNA) wrapped in a vector and producing transient expression of disease-specific CAR or TCR. B: Describe how to make nanoparticles. C: particle size distribution, measured by NanoSight NS300 instrument.
Firstly, it is determined whether adding targeted IVT mRNA nanocarriers in human lymphocyte culture can stably transfect cells. In order to test this technique in clinical related systems, the nanoparticles were loaded with IVT mRNA encoding leukemia-specific 1928z CAR (Figure 3a-e). CD19 targeting receptor is the most researched CAR-T cell product. In the second example, IVT mRNA encoding high affinity HBV-specific TCR is provided here (Figure 3f-j). T cell therapy for chronic hepatitis B is a new method to restore antiviral immunity and cure infection. HBcore18-27 TCR against HBV core antigen was isolated from a HLA-A02.01 donor who had solved HBV infection. For 1928z CAR and HBcore18-27 TCR constructs, real-time quantitative PCR and flow cytometry were used to measure their expression levels in human T cells after transfection with single nanoparticles. It was found that the transgenic expression reached its peak at 24 hours after nanoparticles were exposed, and then gradually decreased (Figure 3a,f). It is worth noting that only nanoparticles functionalized with T-cell-specific antibodies (anti-CD8 or anti-CD3) can effectively deliver transgenes, while the gene expression produced by isotype control functionalized nanoparticles is close to the background level (Figure S1). This translates into a high level of expression on the surface of CAR or TCR, reaching a maximum on the second day.(75% 11% T cells express 1928z CAR, Figure 3b, C; On average, 89 4% of T cells expressed HBcore18-27 TCR (fig. 3g,h). As expected, the receptor expression was transient. After 8 days of culture, the receptor expression of CAR and TCR decreased to 28 6% and 26 9% respectively. Next, the virus method was used to compare the functions (killing and cytokine production) of T cells transfected with nanoparticles and T cells designed with these receptors. In order to prove the specificity of tumor antigen, T cells transduced with CAR gene (P28z, targeting prostate specific membrane antigen) or TCR gene (MSLN-TCR, mesothelin specific) unrelated to tumor were used as control group. Using real-time IncuCyte? living cell analysis, it is impossible to measure the significant difference in the ability of T cells transduced by IVT mRNA to selectively lyse antigen-positive target cells (Raji lymphoma cells in 1928z CAR and HepG2 hepatoma cells in HBcAg stably transduced HBcAg for HBcore18-27 TCR) (Figure 3d,i). In addition, similar levels of effector cytokines secreted by T cells were also detected in the nanoparticle transfection group and the virus transduction group (Figure 3e,j).

Fig. 3 IVT mRNA nanocarriers effectively transfect human T cells by CAR or TCR transgene. Isolated human CD8+T cells were stimulated by beads coated with antibodies against TCR/CD3 and CD28 receptor. After 24 hours, beads were removed, and CD8 targeted nanoparticles (NPs) containing mRNA encoding leukemia-specific 1928zCAR(a-e) or HBcore18-27 TCR(f-j) were mixed into the cell suspension at a concentration of 3 μ 3μg mRNA/10^6 cells. A: QCPR was used to detect the relative 1928z CAR mRNA expression of T cells exposed to 1928z CAR NPs over time. B: T cells were detected by flow cytometry at different time points after incubation with NPs carrying 1928z encoded mRNA. C: Summary chart of gene transfer efficiency in vitro. D: To compare the killing activity of T cells of nanoparticles and retrovirus transfection group on Raji lymphoma cells in vitro. T cells and Raji tumor cells were co-cultured in a ratio of 5:1. The IncuCyte living cell analysis system was used to quantify the immune cell killing of Raji NucLight red blood cells by T cells transfected with 1928z-CAR or control (P28z-CAR) over time. E: The secretion of IL-2(24h), TNF-α and IFN-γ(48h) was detected by e:ELISA.The HBcore18-27 TCR mRNA of f-j is the same.
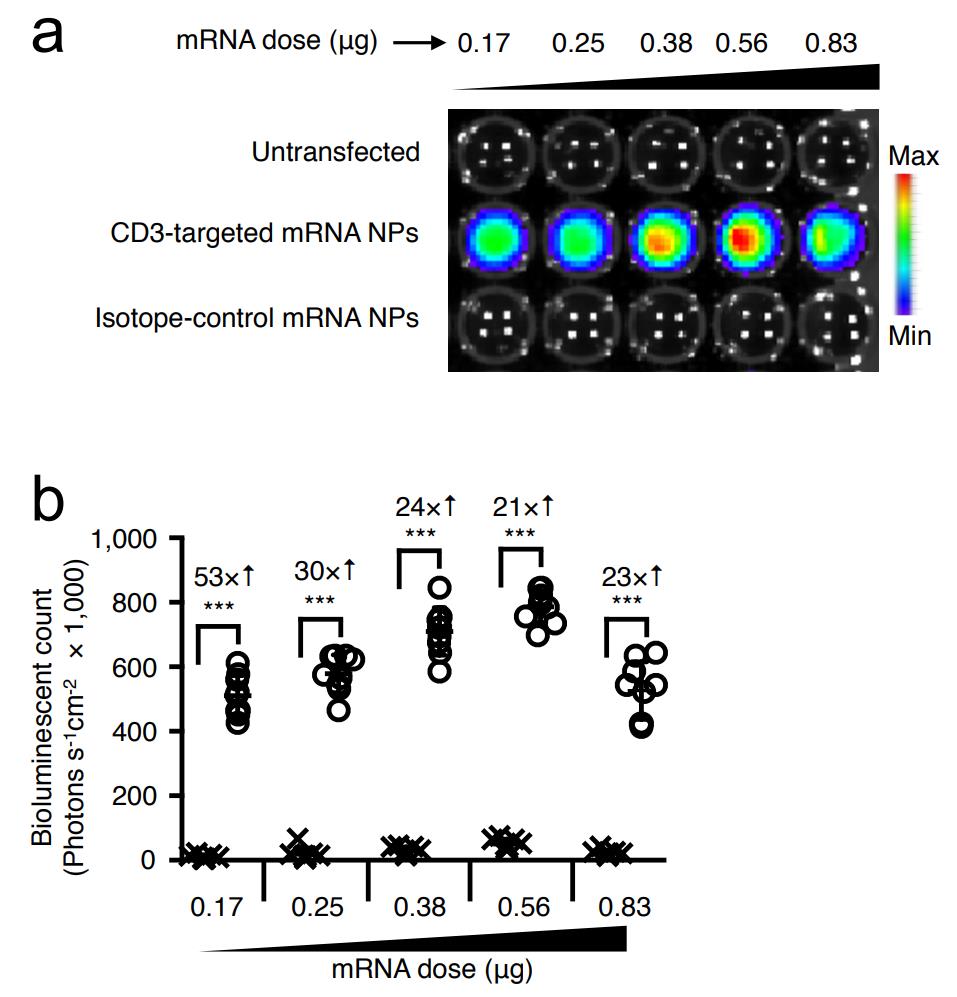
Fig. S1 CD3-targeted mRNA nanoparticles selectively transfect human T cells.
Recognition of leukemia by reprogramming host T cells with nanoparticles
Next, it is studied whether IVT mRNA NPs targeting lymphocytes can reprogram the circulating T cells, and the number is large enough to achieve tumor regression with similar effects as traditional methods. As the first in vivo test system, 1× 10 6 CD19+Raji cells expressing firefly luciferase were inoculated into immunocompromised NOD. CG-PRKDCSCID IL 2RGT M1WJL/SZJ (NSG) mice to establish leukemia model. Five days later, mice were recombined into 10× 10 6 CD3+human T cells, and nanoparticles loaded with the gene encoding 1928z CAR (50μg/ dose) were infused six times a week to produce leukemia-specific or control particles loaded with the mRNA encoding GFP (Figure 4a). According to the kinetics of in vitro measurement of CAR surface expression with IVT mRNA nanoparticles, a weekly administration regimen of nanoparticles was selected, which showed the expression of related receptors for 8 days (Figure 3b,c). In order to compare the efficacy of nanoparticle infusion with traditional adoptive T cell therapy, 5×10^6 T cells were transduced into another group of mice by lentivirus encoding 1928z CAR in vitro. This amount is equivalent to the higher dose of CAR-T cells used in clinical research at present. In clinical research, patients were treated with CAR-T cells weighing as high as 1.2×10^7 CAR-T kilogram. In another dosage regimen,The adoptive transfer of lentivirus-transduced CAR-T cells was combined with systemic injection of nanoparticles carrying control GFP mRNA to determine whether nanoparticle-mediated transfection damaged the anti-tumor function of T cells. The mice in the control group were either not treated or infused with untransformed human effector T cells. Tumor growth was continuously quantified by bioluminescence imaging and the difference of survival rate was monitored. It was found that the adoptive transfer of 1928z CAR-T cells engineered in vitro significantly improved the survival rate. Of the 10 mice, 6 tumors were eradicated, and the tumors of the other mice subsided, and the average 32-day survival rate increased (Figure 4b,c). This therapeutic benefit obtained by traditional adoptive T cell therapy is similar to the treatment of IVT mRNA nanoparticles which programmed the same CAR into lymphocytes in vivo, which achieved the eradication of 7 tumors in 10 mice and the average survival time of recurrent animals was 37 days (Figure 4c). Flow cytometry analysis of peripheral blood 2 days after the first administration showed that nanoparticles carrying 1928z effectively reprogrammed circulating T cells to identify leukemia cells (average 10% 4.3% CAR+CD8+, Figure 4d,e). As expected, the transient expression of these CARs lasted for one week (0.8 0.4% of CAR+CD8+T cells on the 7th day). It is worth noting that repeated doses of nanoparticles are as effective as the first injection, with an average of 10.7 3.6% gene transfer to host T cells (Figure 4e). This shows that,Although IVT mRNA is transient, it can be used as a platform for continuous in situ CAR expression in host lymphocytes.
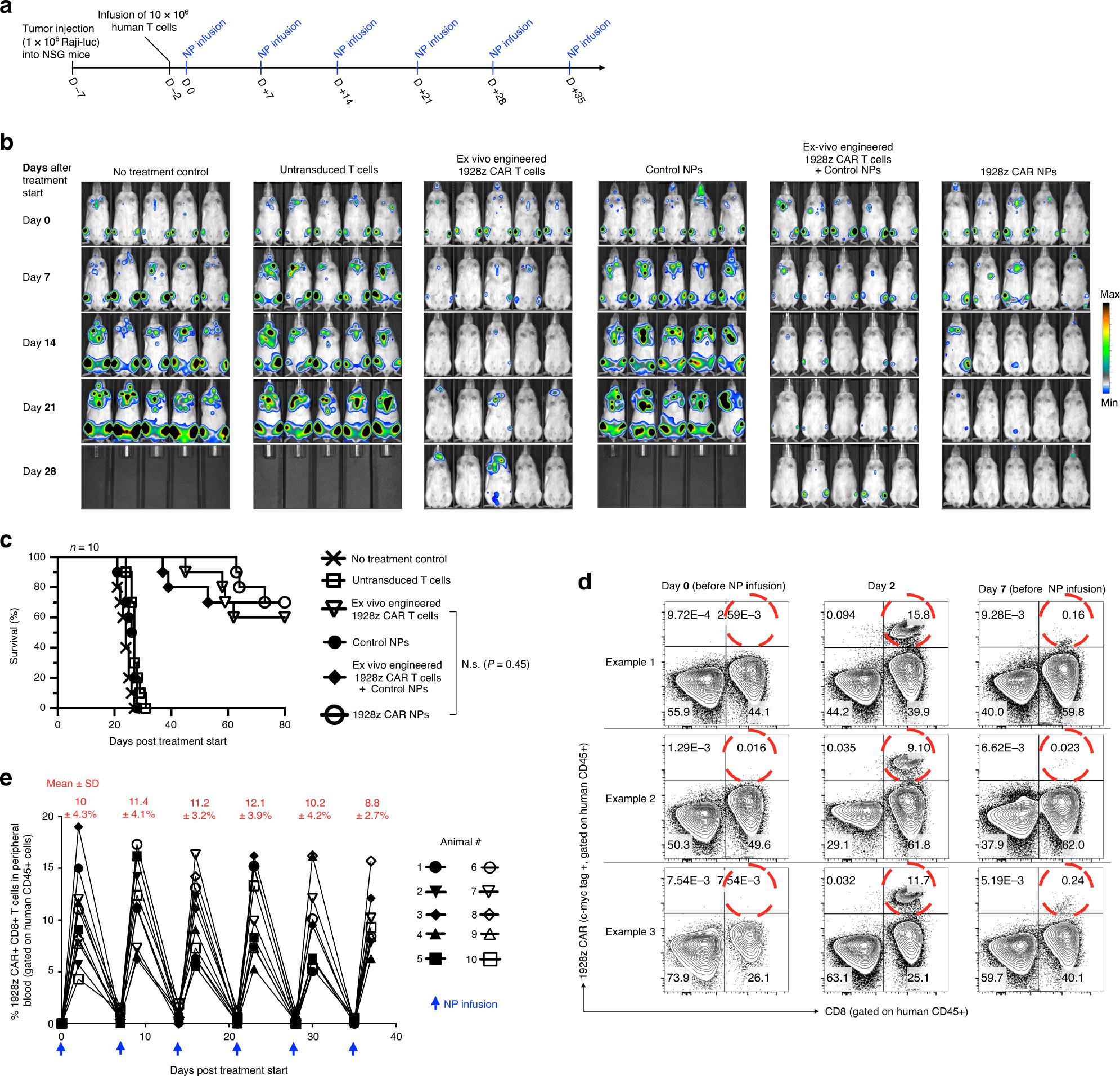
Fig. 4 CAR lymphocytes programmed with nanoparticles can lead to leukemia regression, and its curative effect is similar to adoptive t cell therapy. A: Timeline and administration scheme of nanoparticles (NP). B: sequential biological imaging of Raji lymphoma cells expressing firefly luciferase injected into NSG mice. C: The survival rate of animals after treatment is depicted as Kaplan-Meier curve. D: Flow cytometry of peripheral T cells before and after injection of nanoparticles carrying IVT mRNA encoding 1928z CAR. E: shows the percentage of CD8+T cells transfected by CAR after repeated infusion of 1928z CAR NPs. Each line represents an animal. The average transfection rate (SD) at each time point is displayed at the top.
Therapeutic response of hosts with normal immune function
In order to study how exclusive targeting limits the interaction of nanoparticles to circulating T cells and how it affects their fate, Ai14 reporter mice with complete immune activity were used. In this transgenic model, all cells contain a termination box flanking loxP, which prevents the transcription of tdTomato protein driven by CAG promoter. Only the cells that successfully transduced the mRNA encoding Cre recombinase (Cre) would cut off the termination cassette flanking loxP, resulting in permanent tdTomato transcription and then strongly amplified tdTomato expression. Firstly, the fluorescence of the whole organ in Ai14 mice was measured after injecting CD3-targeted (or isotype-controlled functionalized) nanoparticles carrying Cre mRNA. The gene expression mediated by non-targeted particles is the highest in the liver, while the gene transfer induced by lymphocyte-targeted nanocarriers is mainly in the spleen, lymph nodes and thymus (Figure 5a,b). Detailed flow cytometry analysis of spleen (Figure 5c) showed that CD3-targeted nanoparticles preferentially transfected T cells (8.1 1.9%) without affecting the activity. Other CD45+ subtypes, such as macrophages (3.2 1.5%), B cells (1.1 0.9%), neutrophils (0.3 0.2%) and DC cells (1.9 0.8%), have lower dtTomato signal levels.

Fig. 5 Effective T cell targeting in mice with normal immune function. B6. CG-GT (Rosa) 26Sortm14 (CAG-TDtomato) Hze/J (AI Reporter) Mice were injected with 3 doses of nanoparticles containing 15μg mRNA encoding nuclear localization signal (NLS)-Cre intravenously every day. Nanoparticles were targeted to mouse T cells using full-length anti-CD3 MuIgG2a or IgG2a isotype control. Both antibodies were designed as LALAPG variants to eliminate Fc receptor binding and complement activation. 48h after the last injection, the organs were collected and the dtTomato fluorescence of the whole organs was measured by fluorescence IVIS imaging. Single cell suspensions of spleen and blood were labeled with antibodies against various immune cell subtypes and analyzed by flow cytometry. A: The representative dtTomato expression in organs under fluorescence IVIS imaging. B: Quantify the fluorescence signal of each organ. C: It shows the average SD percentage of immune CD45+dtTomato+ cell types in spleen. Macrophages (CD45+, CD11b+, MHCII+, CD11c?, Ly6C?/Low, Ly6G?), B cells (CD45+, B220+), T cells [CD4+T cells (CD45+, TCRβ+, CD4+, CD8-) and CD8+T cells (CD45) were detected.(CD45+, CD11b+, MHCII+, CD11c, Ly6G+) and DC cells (CD45+, CD11c+, CD11b, MHCII+).
Based on these distribution studies, it is then tested whether the measured number of mRNA nanoparticles redirected T cells is enough to reduce established cancers in fully immune hosts. To this end, Eμ-ALL01 leukemia cells expressing luciferase were injected into albino C57BL/6 mice (a model of B-cell acute lymphoblastic leukemia was established in mice with normal immune function), and bioluminescence imaging was used to quantify the difference of tumor progression between treatment groups (Figure 6a). Mice either received CD3-targeted nanoparticles to deliver mRNA encoding 1928z CAR of the whole mouse, or received GFP control (see Figure S2 for methods). The third group did not receive treatment. It was found that only infusion of nanocarriers encoding 1928z CAR could effectively control the progress of leukemia (Figure 6b,c). Compared with GFP control group, the tumor load was reduced by an average of 26 times after three weeks of treatment.
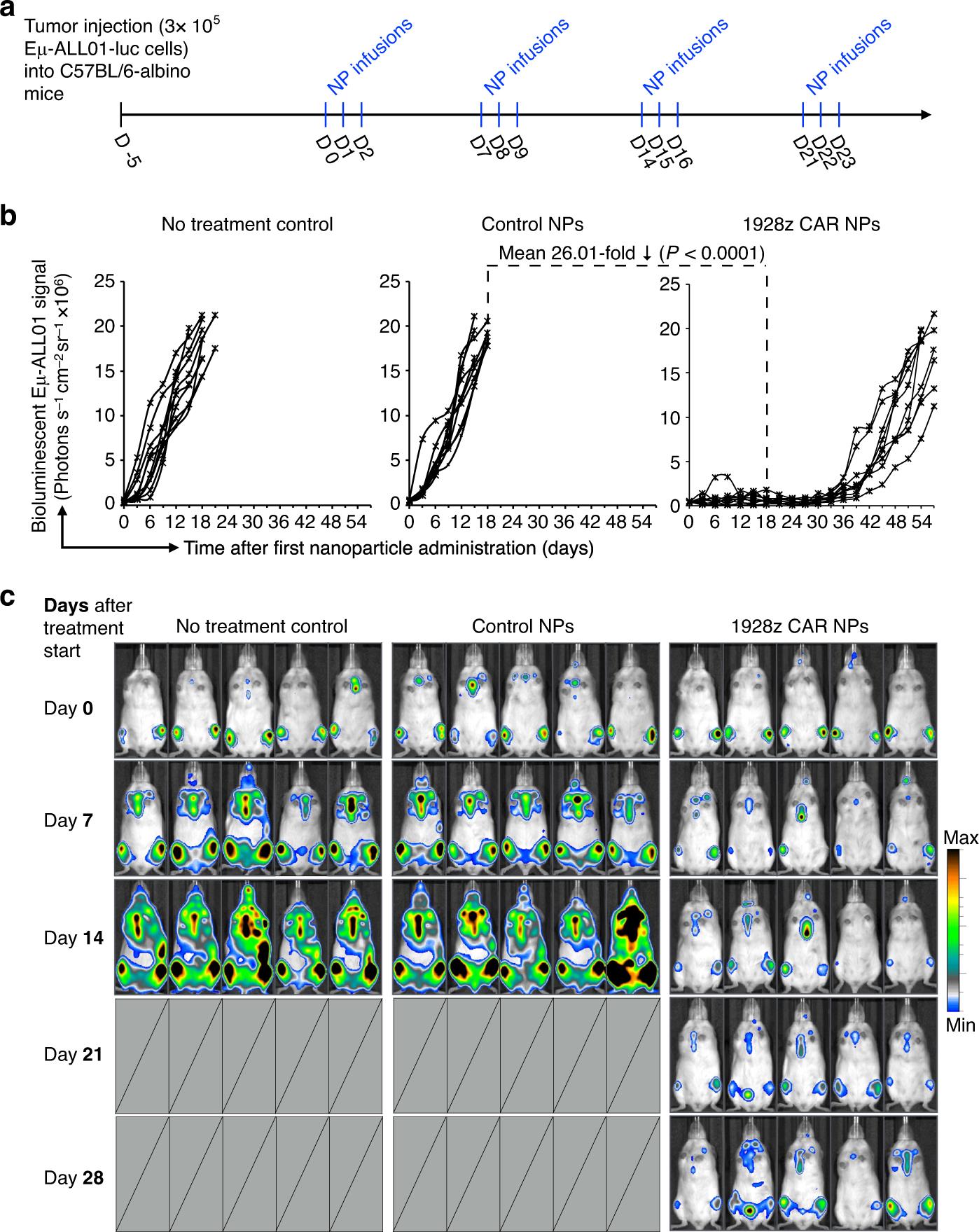
Fig. 6 Anti-leukemia response of mice with normal immune function. A: Timeline and administration plan. B: signal intensity diagram of e-all 01 luciferase after nanoparticle injection. Each line represents an animal, and each point reflects the photon count of the whole animal. C: Sequential biological imaging of Eμ-ALL01 leukemia cells expressing firefly luciferase by systemic injection in albino C57BL/6 mice. Five representative mice from each group (n=10) are shown.

Fig. S2 anti-CD3 mouse IgG2a LALA-PG and non-conjugator control.
Therapeutic response of solid tumor
In order to formalize that this technology is not only related to the treatment of hematological malignant tumors, but also related to the treatment of solid tumors, the ability of nanoparticles aimed at introducing prostate cancer-specific CAR gene into circulating host T cells to induce prostate cancer regression in mice was studied. Unlike leukemia cells, leukemia cells express high levels of CD19 antigen and are easily accessed by circulating lymphocytes, while solid malignant tumors are heterogeneous and protected. This means that some tumor cells will escape the recognition of targeted CAR and will be surrounded by immune suppression defense system, which may lead to T cell dysfunction. In fact, the whole genome/transcription analysis of 140 cases of prostate cancer metastasis was used to determine that prostate cancer lesions showed heterogeneous expression of three key cell surface proteins [prostate specific membrane antigen (PSMA), prostate stem cell antigen (PSCA) and receptor tyrosine kinase-like orphan receptor 1(ROR1)] in patients (Figure 7a). In order to summarize human diseases, LNCaP C42 prostate cancer cells were transplanted into the prostate dorsal lobe of NSG mice in situ (Figure 7c), and the cells showed heterogeneous expression of these key cell surface proteins (Figure 7b). In order to continuously monitor the tumor load by bioluminescence imaging, firefly luciferase (Fluc) was used to mark the tumor cells. After orthotopic transplantation, all the mice developed pathological changes repeatedly within three weeks (Figure 7c, right), and were reconstructed with human 10× 10 6 CD3+human T cells.And randomly assigned to different treatment groups or control groups (fig. 7d). Firstly, the therapeutic effect of systemic injection of 10 6 CAR+T cells transduced in vitro against tumor antigen ROR1 on tumor-bearing mice was tested. It was found that although anti-ROR1 CAR-T cells did not achieve tumor clearance, the survival rate of treated mice more than doubled (69 days compared with 32 days in untreated control group; Fig. 7d). In order to determine whether "ready-made" nano-preparations can achieve similar therapeutic effects, ROR1 CAR transgenic nanoparticles (50μg mRNA/ dose; Fig. 7e). Compared with the untreated control group, particle-induced CAR programming prolongs the survival time by an average of 40 days, which is similar to the survival benefit obtained by traditional adoptive T cell therapy (Figure 7d,f). Proper localization and persistence of T cells is a prerequisite for anti-solid tumor activity, so the frequency of ROR1 CAR-T cells infiltrating into prostate cancer over time was evaluated. The flow cytometry analysis of LNCaP C42 prostate cancer resected on the 4th, 7th and 11th days after T cell metastasis showed that T cells were infused into the tumor site by intravenous infusion (average 892 295 car+T cells /mg tumor tissue), but they could not grow (only 1.04 times the overall expansion between the 4th and 11th days; Fig. 7g,h). In addition, in-situ programmed CAR-T cells effectively penetrated into the tumor (average 648 240 CAR+T cells /mg tumor tissue)., and maintain a high level of CAR transgene (average 91 7 CAR+T cells /mg tumor tissue, Figure 7h) before downregulating the receptor. On the same day, the tumor lesions were infiltrated by freshly reprogrammed peripheral T cells after intravenous injection of ROR1 CAR-encoded mRNA nanoparticles (on the 11th day, the average was 1066 225 car+T cells /mg tumor; Fig. 7h), which summarizes the oscillation dynamics of T cell reprogramming induced by mRNA nanoparticles that have been observed in leukemia research (fig. 4e).
In order to determine the reason why adoptive transferred T cells and injected mRNA nanocarriers failed to completely eliminate the disease, the antigenic phenotype of recurrent prostate cancer was identified by flow cytometry. One of the most common escape strategies in cancer is to reduce the expression of target antigen, because CARs produces selective pressure. In preclinical and clinical studies, this phenomenon is reported as the cause of failure when adoptive T cells targeting only a single antigen are used to treat heterogeneous tumors (such as metastatic prostate cancer). It was found that compared with untreated LNCaP C42 prostate cancer expressing ROR1 tumor antigen at different levels, two treatment groups (adoptive transferred T cells or nanoparticle programmed T cells) finally produced ROR1 low/negative immune escape variants (Figure 7i).

Fig. 7 IVT-mRNA nanocarriers encoding prostate cancer-specific CAR can improve the survival rate of mice with existing diseases. C: Three weeks after transplantation, LNCaP C42 prostate cancer was imaged by bioluminescence in vivo. The picture on the right is a representative photo of prostate dorsal lobe tumor (white arrow). D: Sequential biological imaging of LNCaP C42 prostate cancer cells expressing firefly luciferase transplanted into the prostate of NGS mice in situ. E: Timeline and nanoparticle administration scheme. F: the survival rate of animals after treatment, depicted as Kaplan-Meier curve. G: On the 11th day after treatment, the recovered cells of patients with prostate cancer were detected by multicolor flow cytometry. ROR1 CAR+T cells with adoptive transfer or in situ programming were identified by CD45 and the positive marker of c-myc labeled in the receptor. H: The absolute number of ROR1-CAR+T cells in the tumors isolated on the 4th, 7th and 11th day after the start of treatment. The total number of living cells (trypan blue negative) multiplied by the percentage of ROR1-CAR and CD45 positive. I: Flow cytometry was used to quantitatively detect the expression of ROR1 antigen on LNCaP C42 prostate tumor cells treated with CAR-T cells or ROR1 4-1BBz CAR NP.
HBVIn situ programming of specific T cells
The gene transfer of CARs encoding IVT mRNA can only target T cells to antigens located on the cell surface, so many tumor antigens or virus antigens in cells cannot reach these receptors. It has been proved in vitro that lymphocyte-targeted IVT mRNA nanoparticles can reprogram T cells with engineered TCR, which recognize intracellular HBV core antigen (HBcAg) in HLA background (Figure 3f–J). In view of the fact that more than 300 million people in the world are chronically infected with HBV, and a large number of them develop cirrhosis and liver cancer, it is obviously not feasible to tailor T cell products for each patient. As the first step to treat this disease with IVT mRNA technology, a mouse model of hepatocellular carcinoma (HCC) induced by HBV was established. After laparotomy, 1 million HepG2 cells stably transduced with HBcAg and luciferase were injected into the liver. All mice developed multifocal lesions repeatedly within 7 days (fig. 8a), when they were reconstructed with unstimulated 10× 10 6 CD3+human HLA-A*02:01 T cells, and received twice weekly infusion of nanoparticles loaded with mRNA encoding HBcore18-27 TCR (50μ g/dose) to produce HCC-specific or control particles loaded with mRNA encoding GFP. The mice in the third group were treated with a single dose of 10×10^6 CD3 T cells (HLA-A*02:01).The cells were transduced in vitro with retrovirus vector encoding HBcore18-27 TCR, and the control mice did not receive any treatment. Four days after the second nanoparticle administration (18th day), the liver was isolated to directly quantify the tumor load by bioluminescence imaging, and the single cell suspension was labeled with HBV C18-27 MHC I Pentamer to quantify the percentage of TCR-T cells expressing HBCore 18-27. It was found that enough HBcore antigen-specific T cells were programmed by nanoparticle injection to induce disease regression, and similar therapeutic effects could be achieved compared with engineered lymphocytes in vitro (compared with untreated controls, the photon count was reduced by 13 times and 18.9 times, respectively, Figure 8b,c). Flow cytometry of dissecting liver confirmed that the density of HBcore18-27 TCR-T cells in animals treated with engineered T cells in vitro was equal to that of in-situ programmed nanoparticles (Figure 8d,e).
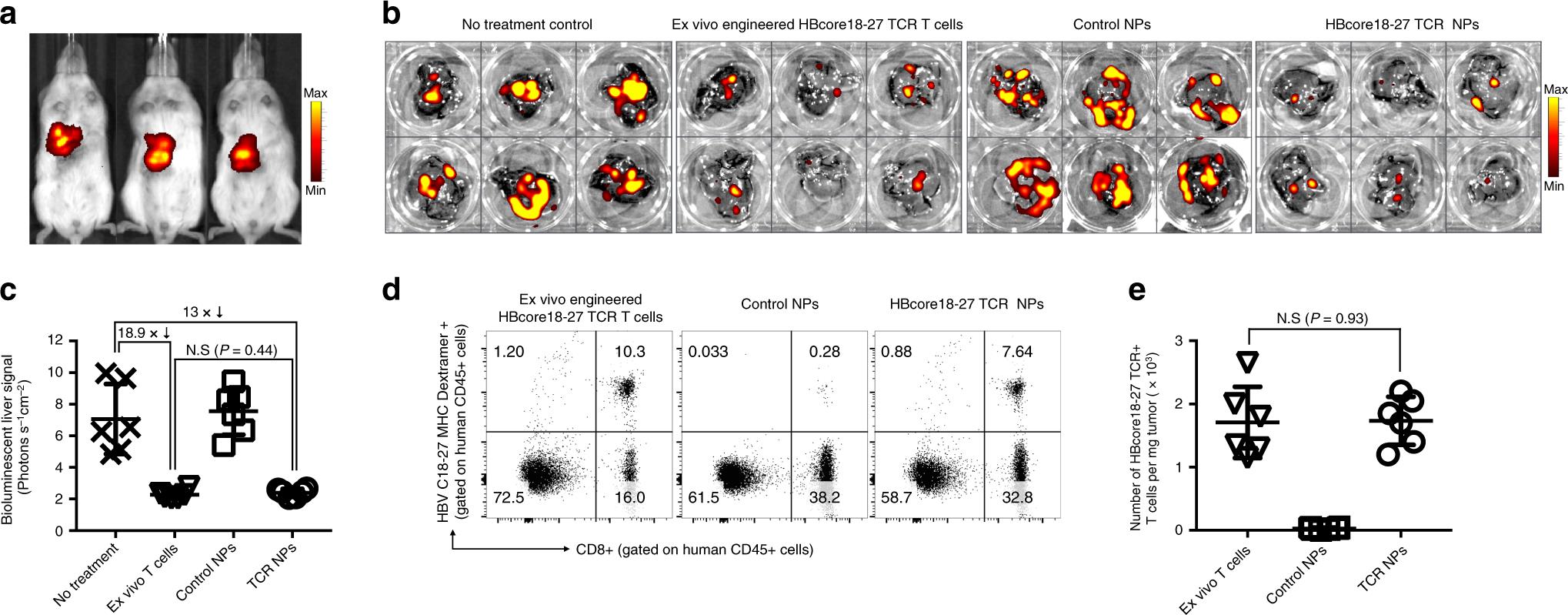
Fig. 8 In-situ programming of HBV-specific T cells using nanoparticles loaded with TCR transgenes. A: A xenograft tumor model of HCC mice induced by HBV was established. HepG2 cells stably transduced with HBcAg and luciferase were injected into the liver of NSG mice reconstructed with human T cells by surgery. Three weeks after implantation, HepG2 tumor was observed by bioluminescence imaging in vivo, and it was assigned to the nanoparticle group (50μg of mRNA encoding HBcore18-17 TCR six times a week) or the T cell treatment group (5×10^6 T cells were transduced with lentivirus encoding HBcore18-17 TCR mRNA vitro). B, c: quantification of bioluminescent liver signal after 6 weeks of treatment. D: Multicolor flow cytometry for recovering cells from the liver 18 days after the start of treatment. The adoptive transfer or in situ programming of HBcore18-27 TCR+ T cells were identified by the positive markers of CD45, CD8 and MHC Pentamer. The absolute number is shown in E. The total cell count of living cells (trypan blue negative) is multiplied by the percentage of HBcore18-27 TCR++,CD8+ and CD45+.
In a word, the results show that repeated infusion of T cell targeting polymer nanocarriers can deliver tumor-specific CAR or virus-specific TCR transgenes to a sufficient number of host T cells, and induce disease regression at a level similar to that of in vitro engineering lymphocyte mass injection.
TCell-programmed nanoparticles are biocompatible
Systematic administration of nano-drugs may cause infusion reaction in patients, which usually delays or stops clinical transformation. These reactions can be manifested as fever, chills, stiffness, rash, chest pain or dyspnea, and in rare cases, they can be fatal. Identifying the risk of infusion reaction early in the process of drug development is helpful to alleviate potential safety concerns after the product enters clinical trials, save time and money for developers, and avoid potential dangerous complications for patients.
The detection cascade scheme developed by NCL is used here, which can indicate the infusion reaction. Specifically, the influence of nanoparticles on complement activation (NCL method ITA-5.2), its hemolysis characteristics (ITA-1) and its influence on T cell oxidative stress (ITA-32) were analyzed. In order to study these effects of nanoparticle concentration in clinical correlation, the theoretical plasma concentration (TPC) was first calculated, which is an effective mouse dose (in the experiment: 50μg mRNA/ dose), and scaled to the equivalent human dose (=2.03μg mRNA/mL blood; Fig. 9a). In order to evaluate the effect of T-cell-targeted mRNA nanoparticles on red blood cells, the hemolysis test was carried out by measuring the release of hemoglobin by spectrophotometry after exposure to different concentrations of particles. The performance of hemolysis test was tested by negative (PBS) and positive (Triton-X) controls. It was found that the hemolysis rate of TPC particles was lower than 2% (the average was 1.21 0.26%, while that of PBS control was 0.7 0.11%; Fig. 9b), which is defined as non-hemolytic. Nano-particles also did not induce the activation of complement iC3b or Bb, while C4d was slightly higher than twice the determination threshold at TPC concentration (average 2.3 0.13%, compared with 1 0.003% in PBS control; Fig. 9c). Finally, mitochondrial oxidative stress was measured as the key determinant of nanoparticle-induced damage, because reactive oxygen species (ROS)Over-production will cause damage to organelles and DNA, and eventually lead to cell death. In addition, another consequence of ROS overproduction is the activation of cell signaling pathways that stimulate the expression of pro-inflammatory and fibrotic cytokines. Compared with PBS control, T-cell-programmed nanoparticles only induced a very mild increase in oxidative stress (average 3.6 0.2 times) (Figure 9d).
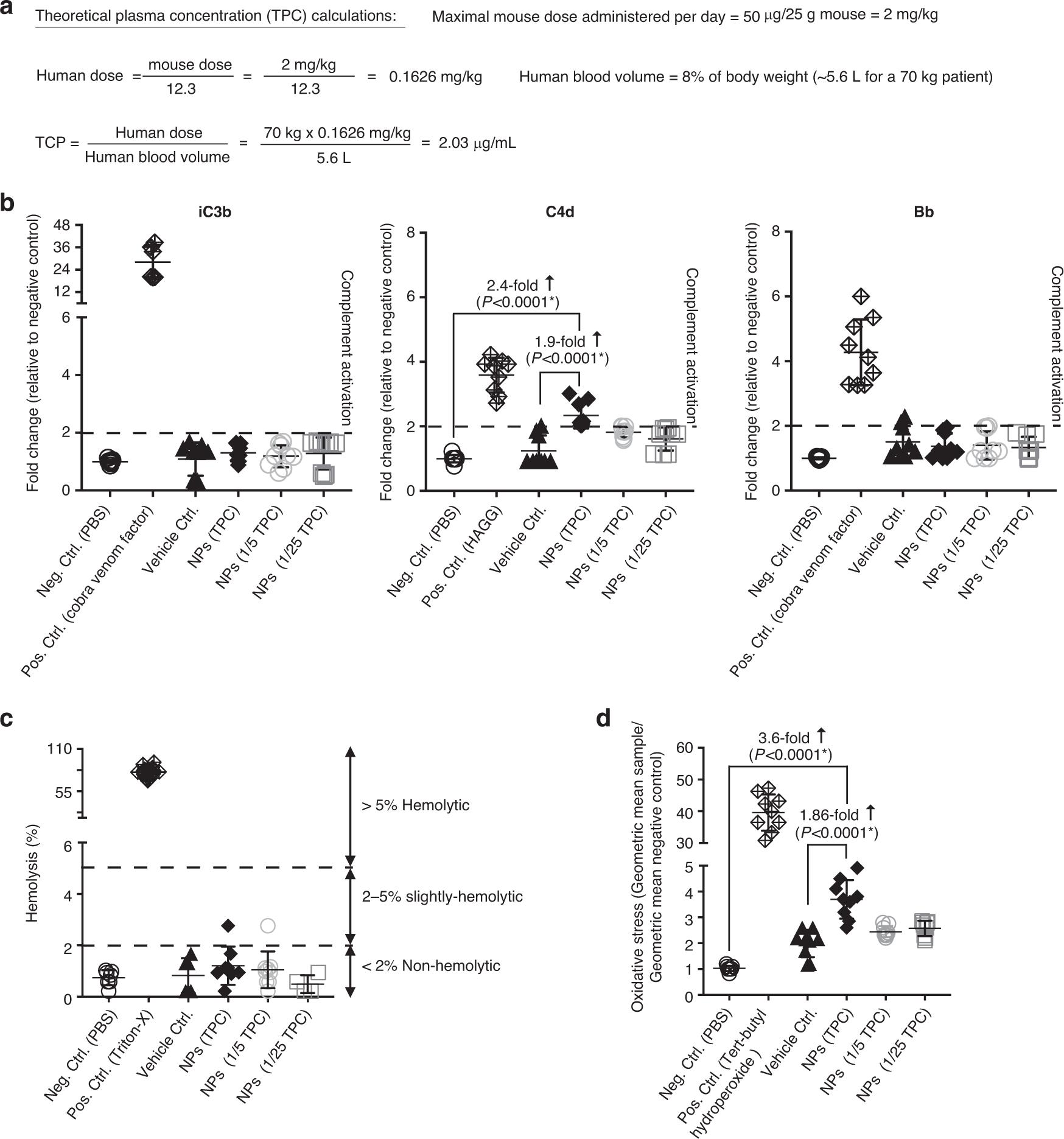
Fig. 9 In vitro analysis of possible infusion reactions. A: calculation of theoretical plasma concentration. Hemolytic activity of b: t cells targeting mRNA nanoparticles. C: quantitative determination of complement activation by enzyme immunoassay. The 2-fold change relative to the negative PBS control is defined as the determination threshold (dotted line). Study on oxidative stress response of lymphocyte mitochondria after d: NP transfection.
Under the guidance of in vitro evaluation of the possible infusion reaction caused by T cell programming mRNA nanoparticles, a comprehensive toxicity assessment was conducted for rodents. Rats are the first rodent species to predict the toxicity of nucleic acid-based molecular therapy to human health, because their metabolic physiology (especially kidney and liver functions) is closer to humans than mice. SD rats (6-8 weeks old) were injected with a dose of nanoparticles carrying 100μg mRNA, which is equivalent to the rat dose of 50μg mRNA in mice, based on the standardization of body surface dose. These experiments were carried out using 1928z CAR nanoparticles. 1928z CAR recognizes human CD19, but does not cross-react with rat CD19 to ensure that the change of measured parameters can be attributed to nanoparticles rather than their reprogramming activities. The control group was injected with 25mM sodium acetate buffer (carrier control group) or not. After 48 hours, the animals were killed, blood samples were taken to determine clinical biochemical indexes, and gross anatomy was carried out. The following tissues were evaluated by pathologists certified by the Committee: lung, liver, heart, brain, kidney, spleen, bone marrow and duodenum. There were no histological changes attributable to nanoparticle drug therapy (fig. 10a). The few lesions noted are mild to mild, which are considered accidental and have nothing to do with the study. Two of the five rats in all groups had the least inflammatory infiltration in the liver. These tiny infiltrations are considered as background lesions and have nothing to do with treatment. Similarly, all groups have individuals with mild to mild chronic inflammation of renal pelvis.The chronic nature of the lesion is inconsistent with the acute treatment effect, so it is considered accidental. Compared with the control group, the whole blood platelet count of the animals in the nanoparticle treatment group was slightly lower (407 115 k/μ l and 290.4 56.3 k/μ l respectively; Fig. 10b). Compared with the control group, the animals in the nanoparticle group also had slight hypoglycemia (average 45.4 26 mg/dl and 78.3 69.8 mg/dl, respectively; Fig. 10c). All other serum chemical indexes (including liver function and renal function) of the rats treated with nanoparticles were similar to those of the control group, indicating that no systemic toxicity occurred. Serum IL-6 level moderately increased to an average of 16.5 pg/ml 5.9 pg/ml (Figure 10d), which can be considered as safe according to previous reports.
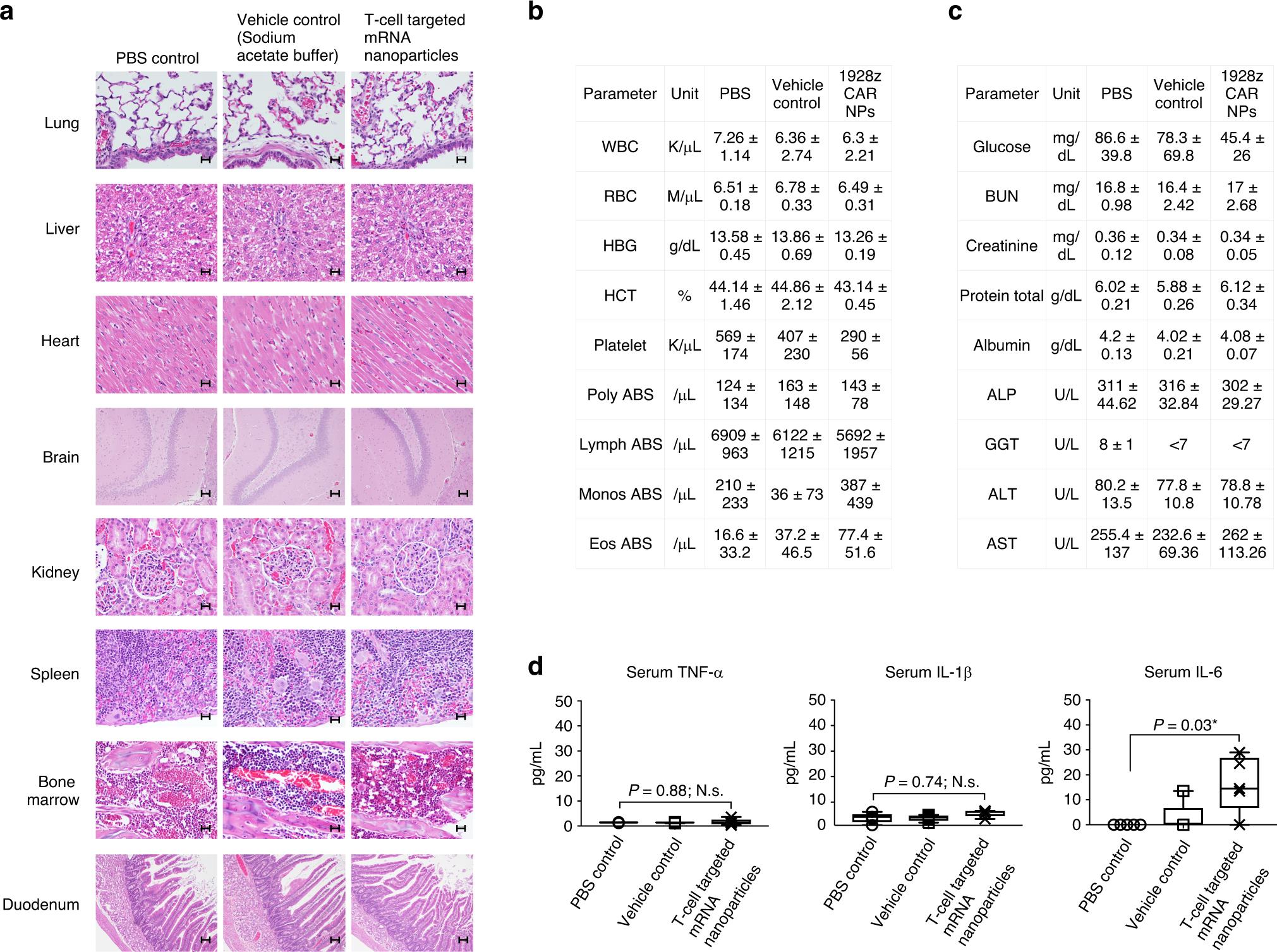
Fig. 10 The infusion of nanocarriers has nothing to do with acute systemic toxicity. Female SD rats were intravenously injected with CD8-targeted IVT mRNA encoding 1928z CAR. After 48 hours, a pathologist certified by the Committee made a comprehensive histopathological evaluation and serum chemical analysis in a blind way. A: representative H&E stained sections of various organs isolated from control or nanoparticle-treated animals. B: blood cell count. C: serum chemistry. D: Detection of serum TNF-α, IL-1β and IL-6 cytokines.
summary
Although T cells modified by CAR and TCR have transformed a few hematologic cancer, it is obvious that their current clinical application only represents a small part of the possibility that this technology may provide. Theoretically, therapeutic T cells can treat malignant tumors and chronic infections with targeted antigens, as proved by a large number of preclinical reports. However, at present, the method of producing disease-specific T cells in vitro is very complicated and cannot support the treatment of a large number of patients, because a new lymphocyte cluster must be produced for each patient. In order to make it easier for patients to obtain T cell products, the field has turned to allogeneic technology to provide a larger scale and lower cost. Several clinical T cell companies have begun to test CAR-T cells, which are "ready-made" products made by healthy unrelated donors rather than patients. Although this method can treat cancer patients who can’t make autologous T cells because of their low lymphocyte count or poor T cell quality, it requires several extra cell engineering steps to prevent donor cells from attacking the host, which in turn prevents patients’ own T cells from rejecting the infusion products. This is usually done by multiple gene editing to remove natural TCR and HLA molecules from T cell products. However, these extra operations increase the complexity, time and cost of the manufacturing process, while reducing the cell yield and vitality. In order to prevent rejection, patients who receive universal CAR-T cells first severely suppress immunity through lymph depletion chemotherapy.This takes time and exposes patients to additional toxicity. Therefore, in vitro engineering of allogeneic cell products is unlikely to significantly increase the number of patients receiving T cell therapy, especially those patients with infectious diseases who need rapid intervention to keep the endogenous immune system intact.
Previously, an injectable DNA-based nano-reagent was described, which can program circulating T cells with leukemia-specific CAR transgene. In order to overcome the inherent low gene transfer of plasmid DNA, plasmid DNA must enter the nucleus to be transcribed into mRNA. The transposon/transposase system encoding CAR was loaded on nanoparticles, and the system was randomly inserted into the genome of the target cell. Although this study provides proof of concept that it is possible to program CAR-T cells in situ with injectable nano-reagents, it will be challenging to transform this DNA nano-drug into clinic for the following reasons: ① Unpredictable genotoxicity and expression kinetics. These nanoparticles stably integrate their therapeutic CAR transgenes into target cells, resulting in permanent genomic changes and unpredictable genotoxicity of various cell types. In addition, once nanoparticles are injected into patients, doctors cannot control the kinetics of CAR expression in vivo; ② The copy number of CAR gene related to each nanoparticle is low. The number of CAR genes that can be loaded into these DNA nanoparticles is limited by the size of the vector skeleton and promoter sequence, and the requirement of stable integration of transposase expression vectors. This greatly limits the efficiency of in-situ gene transfer, especially when trying to deliver large transgenes encoding TCRα and β chains; ③ Abundant tumor antigens are needed to expand the small population of in situ transfected CAR-T cells to the number related to treatment. This amplification period takes time,This is a disadvantage for patients with rapidly progressive diseases or definite solid tumors.
Here, we explore the use of IVT mRNA to quickly and specifically program antigen recognition ability into circulating T cells as a strategy to treat cancer and infectious diseases. Compared with DNA nanocarriers, the synthesized mRNA molecules can be directly translated into therapeutic target proteins without entering the nucleus, thus ensuring high transfection rate and rapid therapeutic effect. Their tailoring size (the actual CAR coding or TCR coding sequence in this study +276 bases of 5′ UTR and polyA region) leads to a high copy number per nanoparticle. In addition, because the delivered mRNA plays its role in cytoplasm, uncontrolled insertion mutation and promoter dependence are avoided. It is proved here that simply injecting well-designed mRNA nanocarriers can selectively introduce CAR gene or TCR gene into host T cells, and program them to make the number sufficient to cause disease regression, which is similar to the adoption method. Several ongoing clinical trials are testing the repeated infusion of in vitro engineered mRNA CAR-T cells (NCT01355965, NCT01897415, NCT02277522 and NCT02624258) in cancer patients, and the first data shows that the instantaneous CAR expression after cell infusion is enough to trigger an anti-tumor response.
Three important reasons why IVT mRNA has rapidly become a new adoptive T cell therapy tool are its inherent safety, efficient translation of recombinant protein and its ability to control the pharmacokinetics of treatment, which is similar to traditional small molecule drugs. In fact, the kinetics of T cells expressing CAR measured in mice after multiple doses is similar to the profile of drugs with a definite half-life (Figure 4e). This is in sharp contrast to the rather unpredictable T cell dynamics after the adoptive transfer of engineering T cells. In this case, the cell concentration in the blood rises to the highest, and then decreases within a variable period of several days to several months. Although in-situ programming has obtained the ability to control the pharmacokinetic characteristics of the therapy and reprogram the fresh population of host lymphocytes regularly, thus potentially bypassing some major obstacles in the wide application of T cell therapy (such as T cell failure and dysfunction, and long-term toxicity), this technology still has some limitations: (1) It depends on the existence of a sufficient number of functional T cells in patients. Lymphocytosis is common in patients with advanced cancer who receive a large number of chemotherapy drugs. Therefore, the patient’s blood is likely to need to be pre-screened before the clinical trial of ready-made nano-reagents. (2) The efficacy of drugs can be passivated by immune response. Because T-cell programmed nanoparticles are regularly injected into patients with intact immunity, anti-drug antibodies may be formed. For the clinical transformation of this technology, it is important to select a fully humanized CD8 targeting ligand.To provide CAR/TCR structure with low immune risk, and synthesize mRNA with pseudouridine (or N1- methyl-pseudouridine described recently) and 5- methylcytosine to reduce innate immune response.
In order to redirect circulating T cells to resident tumor cells in situ, several biotechnological drug manufacturers have developed bispecific antibodies, including BiTEs, DART and diabodies. Among them, blinatumomab (a CD19-specific BiTE) has shown encouraging results in the clinical study of patients with hematological malignancies. However, BiTEs must be continuously infused, which will produce systemic toxicity. In addition, like traditional monoclonal antibodies, BiTE does not undergo active biological distribution or self-expansion after infusion. In contrast, the gene modification system based on nanoparticles described here can produce new tumor-specific T cells, which, as a "living drug", actively locate at the target, increase in number and continuously destroy cancer cells. People’s interest in CAR-T cell therapy is still strong, so it is time to introduce ready-made nano-reagents as a competitive technology, which can quickly reprogram T cells, identify and destroy tumors without laboratory operation.
Before conducting clinical research on humans, we will refer to the FDA’s regulations on nano-drugs and expand cooperation with NCL to confirm the safety of nano-particles in large animal species. Different from the treatment methods established in clinic (such as small molecules or antibodies), CAR-programmed nanoparticles are multi-component three-dimensional structures, which require repeatable manufacturing processes to reliably realize the expected physical and chemical characteristics, biological behavior and pharmacological characteristics. The safety and effectiveness of this nano-drug may be affected by small changes in many parameters, which need to be carefully monitored, especially in the case of targeting unexpected sites and potential toxicity. In addition, compared with traditional drugs, nano-drugs need additional development and regulatory considerations.
Richards, M. FDA approves first cell-based gene therapy for adult patients with relapsed or refractory MCL. (2020).
Curran, E. & Stock, W. Taking a “BiTE out of ALL”: blinatumomab approval for MRD-positive ALL. Blood 133, 1715–1719 (2019).
Mueller, K. T. et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 130, 2317–2325 (2017).
Beatty, G. L. et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 155, 29–32 (2018).
Foster, J. B., et al. The emerging role of in vitro-transcribed mRNA in adoptive T cell immunotherapy. Mol. Ther. 27, 747–756 (2019).
Calmes-Miller, J. FDA approves second CAR T-cell therapy. Cancer Discov. 8, 5–6 (2018).
Parayath, N.N., et al. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun 11, 6080 (2020).
Smith, T. T. et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 12, 813–820 (2017).
Kah, J. et al. Lymphocytes transiently expressing virus-specific T cell receptors reduce hepatitis B virus infection. J. Clin. Invest. 127, 3177–3188 (2017).